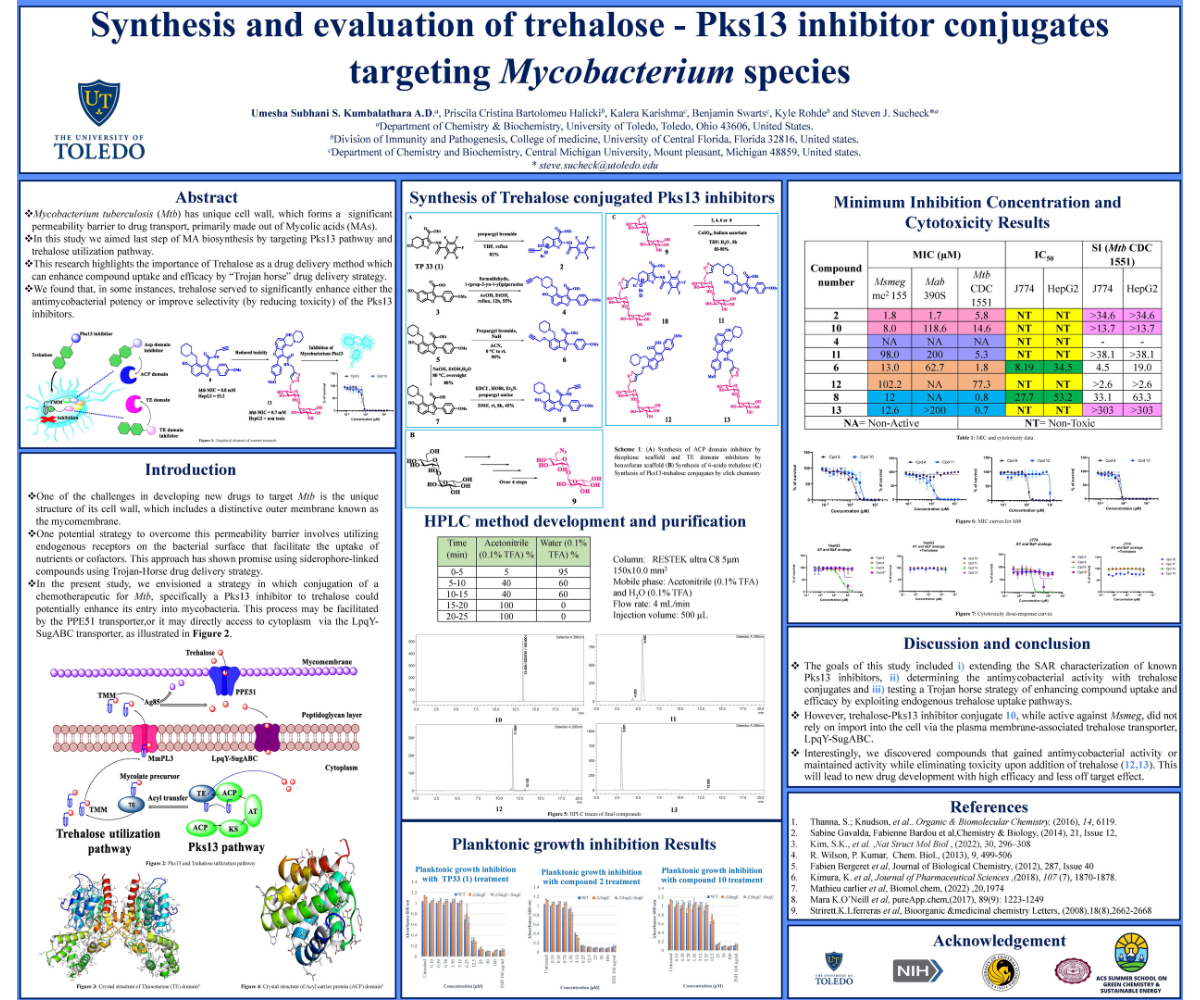
Mycobacterium tuberculosis (Mtb), an opportunistic pathogen, is responsible for tuberculosis (TB), one of the deadliest infectious diseases. The mycomembrane of Mtb contributes to its resistance against many broad-spectrum antibiotics due to its lipid-rich structure, primarily composed of mycolic acids. Inhibiting the biosynthesis of mycolic acids is a promising strategy in TB drug discovery, as this pathway is enriched with essential enzymes that could serve as selective drug targets. This study focuses on the last step of mycolic acid biosynthesis, catalyzed by Polyketide Synthase 13 (Pks13), which forms α-alkyl-β-keto esters by condensing fatty acyl chains. Trehalose, a natural disaccharide, is involved in transferring mycolate precursors to the periplasmic space via specific transporter systems, contributing to cell wall integrity and formation of cord factors in this pathway.
The biochemical relationship between the Pks13 and trehalose pathways remains poorly understood. To bridge this gap, we utilized a green chemistry approach by conjugating azide-modified trehalose analogs with novel Pks13 inhibitors through a "trojan horse" drug delivery strategy, enabling safer, more efficient targeting of the enzyme complex. We selected two key domains for inhibition: the Acyl Carrier Protein (ACP) domain and the Thioesterase (TE) domain. The ACP domain inhibitor was synthesized using a sustainable, one-step Gewald reaction, while the TE domain inhibitors were synthesized via a green Mannich reaction and EDC-mediated coupling with mild and environmentally friendly purification techniques.
We first evaluated the Minimum Inhibitory Concentration (MIC) of the AT and BzF analogs, along with their trehalose conjugates, against Mtb and two non-tuberculosis species, using the trojan horse strategy. Toxicity was assessed against macrophages and hepatocytes to evaluate the safety profile of the compounds. In addition, we investigated the uptake of these compounds via the Lpqy-SugABC transporter system, known for importing trehalose and its analogs. The compounds demonstrated species-specific antimicrobial activity, with potent effects against Mtb, Mycobacterium smegmatis (Msmeg), and Mycobacterium abscessus (Mab). Remarkably, one BzF-trehalose analog displayed enhanced antimicrobial activity while eliminating cytotoxicity, demonstrating the promise of this approach.
Our findings suggest that conjugating drug-like molecules to naturally occurring trehalose can enhance antimicrobial efficacy while minimizing cytotoxicity, offering a sustainable and safer alternative for developing anti-tuberculosis and non-tuberculosis therapies. Additionally, this research supports green chemistry principles by utilizing environmentally benign synthetic methods, reducing waste, and minimizing toxic by-products in drug development. This work may guide future drug discovery efforts, emphasizing sustainable practices and improved safety profiles.